mergem
merging, comparing, and translating genome-scale metabolic models
Summary
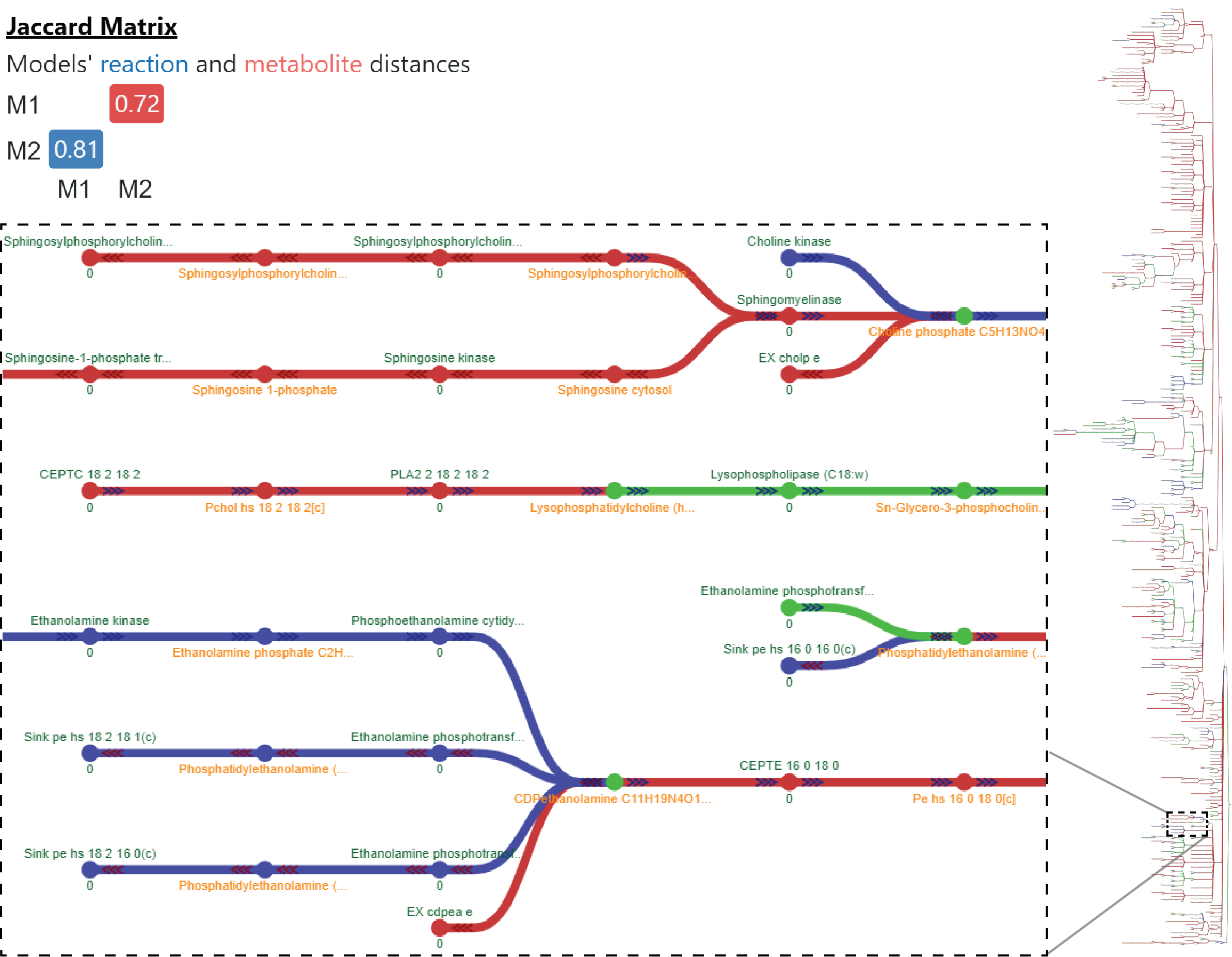
mergem is a free Python library and command-line tool for merging two or more genome-scale metabolic models. The tool can take models in various COBRApy compatible formats such as SBML, JSON, etc. and even COBRApy model objects, when the package is imported. The results of a single merge include the merged model, jaccard distances between all pairs of models, number of metabolites and reactions merged, and lists of models that contain each metabolite and reaction. mergem is also available in the user-friendly application Fluxer, which produces tidy flux graphs that can visually compare the complete metabolic network from multiple models.
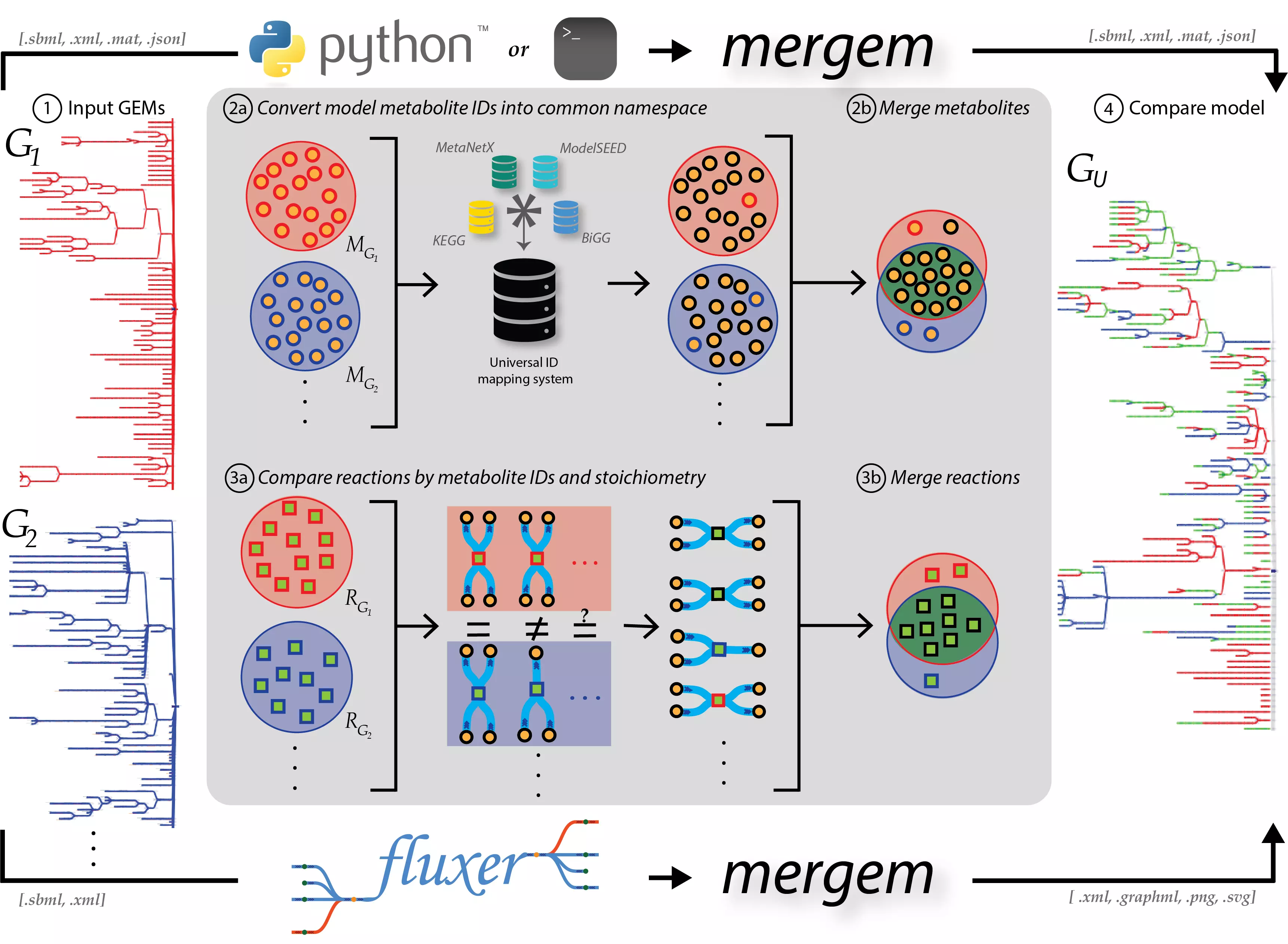
Functionality
For each input model, mergem converts the metabolite IDs into a common namespace using a database ID mapping dictionary. Reactions are compared using the participating metabolites (after conversion to common namespace). The metabolite ID mapping dictionary contains metabolite identifiers from various databases such as ModelSEED, KEGG, ChEBI, and MetaNetX that have been unified per metabolite. The dictionary thus allows for model metabolites to be compared more efficiently. The mapping dictionaries can be updated, during which the latest identifier information is downloaded from each database and identifiers representing the same metabolite are mapped to one another.
Installation, Documentation, and Source Code
mergem is freely available via PyPI and can be pip installed with pip install mergem. Detailed documentation for mergem is available here. A tutorial for using mergem in the user-friendly web application Fluxer can be found on this page. mergem is ditributed under a GPLv3 license and the source code can be obtained from this GitHub repository. If you find mergem useful, please cite the following paper in your work.
Citation
mergem: merging, comparing, and translating genome-scale metabolic models using universal identifiers
A. Hari, A. Zarrabi, D. Lobo
NAR Genomics and Bioinformatics 6(1), lqae010, 2024.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM137953. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.